Hemoglobin S Sickle Cell Anemia
_with_normal_hemoglobin_(hba)_vs_sickled_rbc_wi.png)
What Is Hemoglobin S and How It Causes Sickle Cell Anemia
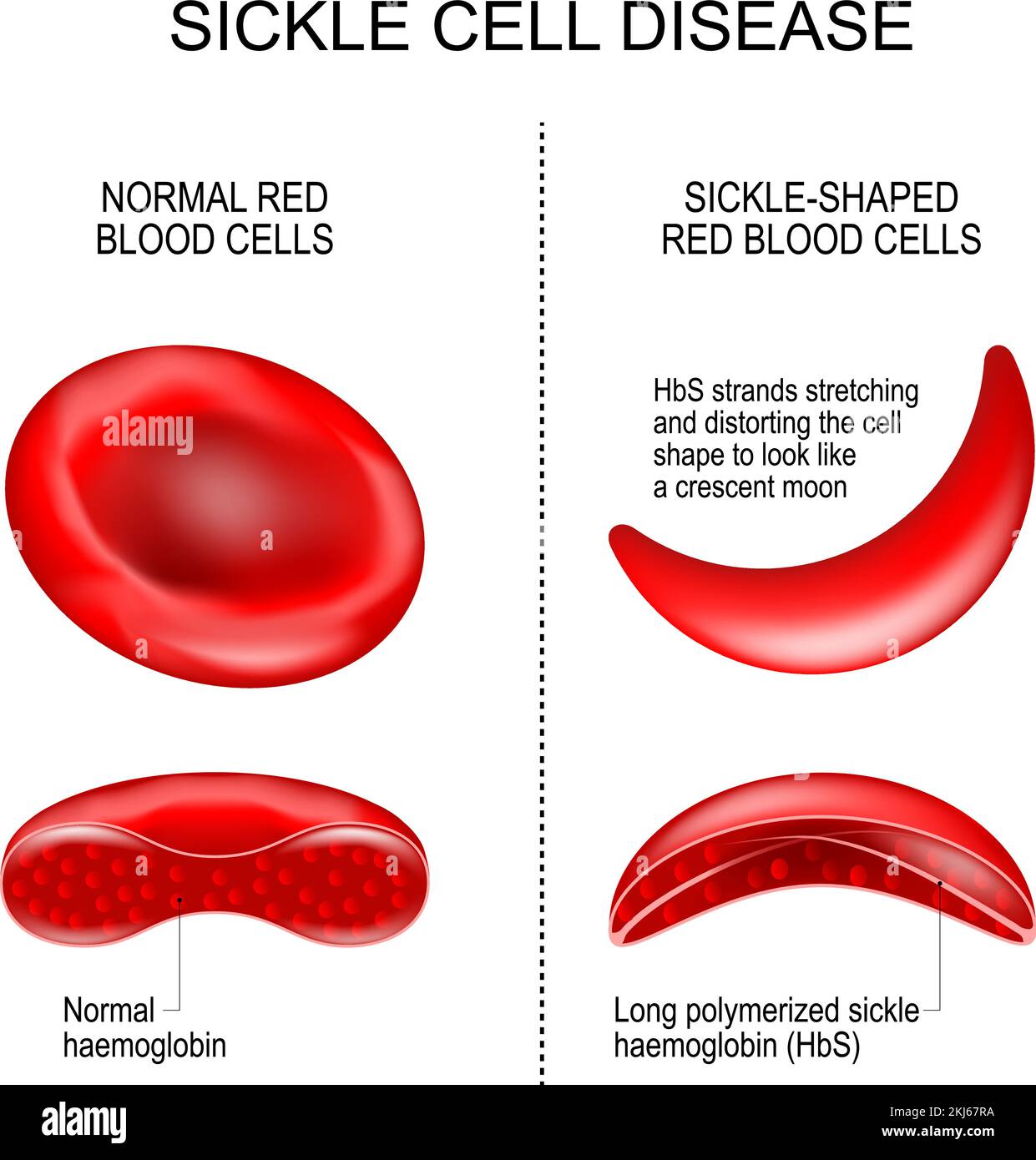
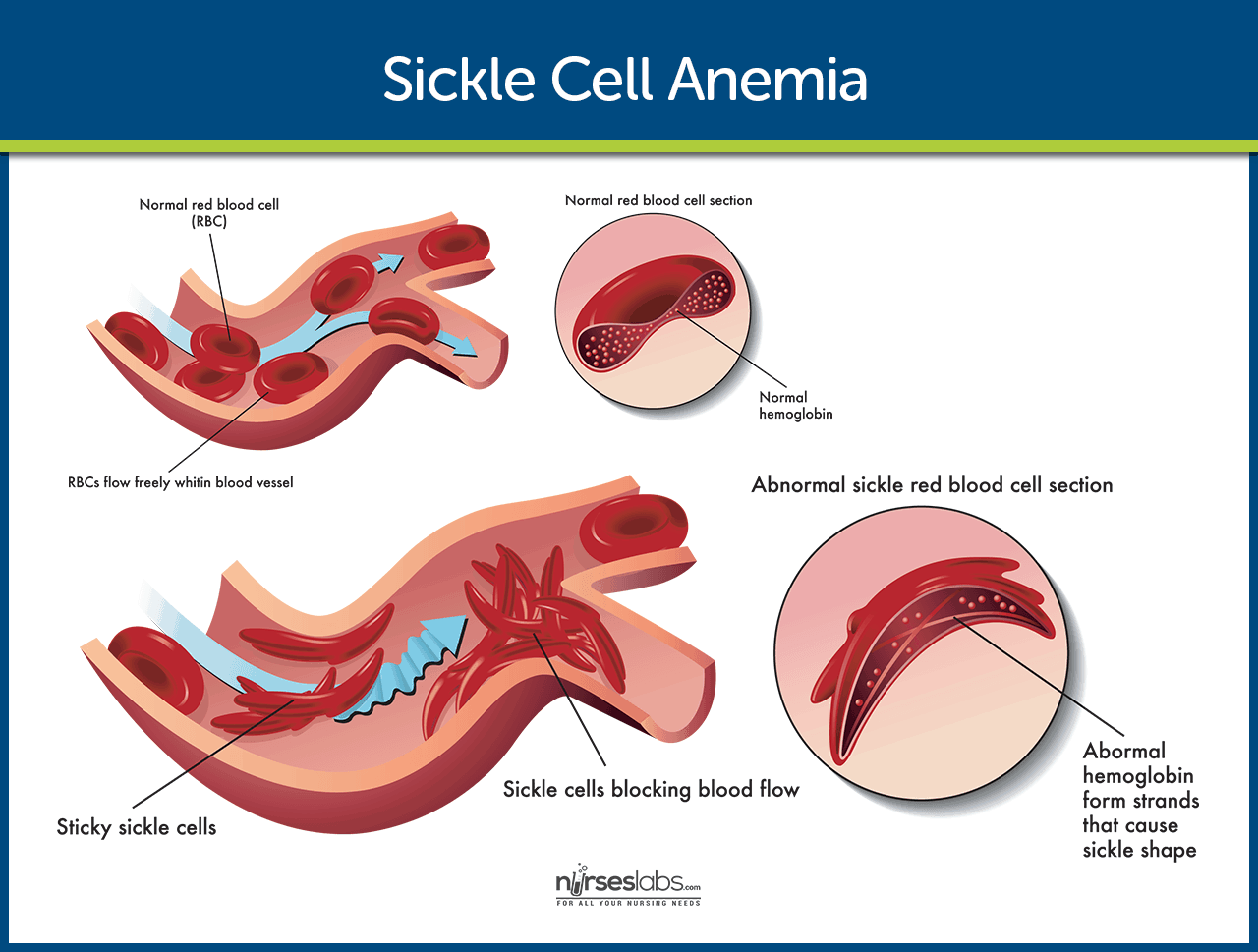
Hemoglobin is the protein in red blood cells that carries oxygen throughout the body. In people with sickle cell anemia, a mutation changes the structure of hemoglobin, producing hemoglobin S. When hemoglobin S releases oxygen, it can stick together and form long, rigid rods that distort the soft, round red blood cells into a sickle or crescent shape.
These sickle shaped cells are fragile and break apart easily, which leads to a constant shortage of red blood cells, or hemolytic anemia. Because they are also stiff and sticky, they can block small blood vessels. This blockage slows or stops blood flow, which triggers episodes of severe pain and can damage organs over time.
Common Symptoms and Early Signs to Watch For
The signs of hemoglobin S related disease often appear in early childhood and vary from person to person. The most common symptoms include episodes of pain, called sickle cell crises, which can last from hours to weeks. Many people also feel tired, pale, and short of breath because of the anemia.

Other frequent symptoms include jaundice, which causes yellowing of the skin and eyes, and swelling in the hands and feet due to blocked blood flow. Children with sickle cell disease may experience delayed growth or frequent infections because the abnormal blood flow affects the spleen and other organs.
- Severe pain in the chest, back, or joints
- Persistent fatigue and weakness
- Frequent infections and fever
- Pale skin or noticeable yellowing
How Hemoglobin S Is Inherited and Who Is at Risk
Sickle cell anemia is inherited in an autosomal recessive pattern, which means a child must inherit two copies of the mutated gene, one from each parent, to have the disease. Parents who carry only one copy are usually healthy but are called carriers. Carriers can pass the gene to their children without showing symptoms themselves.
The condition is most common in people of African, Mediterranean, Middle Eastern, and South Asian descent. In regions where malaria is or was common, having one copy of the sickle cell gene can actually provide some protection against severe malaria. This natural selection is why hemoglobin S traits remain at higher levels in certain populations today.
Diagnosis and Modern Screening Methods
Newborn screening programs in many countries now test for sickle cell disease shortly after birth. A simple blood test checks for the presence of hemoglobin S and other abnormal hemoglobins. Early diagnosis allows families and healthcare teams to start treatment right away, which can significantly improve outcomes.
Prenatal testing is also available for couples who know they are carriers. Options such as genetic counseling, chorionic villus sampling, or amniocentesis can help parents understand their risks and make informed choices. Accurate diagnosis is the foundation for personalized care and long term management.
Treatment Approaches and Daily Management
There is currently no universal cure for sickle cell disease, but several treatments can reduce pain, prevent complications, and improve quality of life. Hydroxyurea is a medication that can increase fetal hemoglobin levels, which helps reduce the production of hemoglobin S and the frequency of crises.

Regular medical care, immunizations, and antibiotics help prevent infections. Folic acid supplements support red blood cell production, and pain management strategies are tailored to each person’s needs. In some cases, blood transfusions or newer therapies like gene therapy may be considered as part of a comprehensive plan.
Lifestyle Adjustments and Long Term Outlook
People with hemoglobin S related disease can lead full and active lives by adopting certain habits. Staying well hydrated, avoiding extreme temperatures, and managing stress can lower the chances of a sickle cell crisis. Simple steps like gentle exercise and balanced nutrition support overall health and energy.
Working closely with a healthcare team helps monitor organ function and adjust treatment over time. Advances in research continue to improve understanding of hemoglobin S and open doors to better therapies. With proper care and support, many individuals with sickle cell disease enjoy meaningful, productive lives.
Conclusion
Hemoglobin S sickle cell anemia is a complex condition caused by a single genetic mutation that changes how red blood cells carry oxygen. Understanding the role of hemoglobin S, recognizing the symptoms early, and accessing modern screening and treatment can make a profound difference. With informed care and a supportive environment, people living with this disease can manage pain, reduce complications, and build a strong, healthy future.
Sickle cell anemia - causes, symptoms, diagnosis, treatment & pathology
What is sickle cell anemia? Sickle cell anemia is an autosomal recessive genetic condition where the beta-globin protein subunit ...
